Gaussian 16
Gaussian 16 to najnowsza wersja pakietu programów zajmującego się strukturą elektronową, używanych przez chemików, inżynierów chemików, biochemików, fizyków i innych naukowców na całym świecie.
Aplikacja zapewnia szeroki pakiet najbardziej zaawansowanych dostępnych możliwości modelowania. Można go używać do badania rzeczywistych problemów chemicznych w całej ich złożoności, nawet na skromnym sprzęcie komputerowym.
Dzięki Gaussian 16 można dokładnie zbadać problemy chemiczne. Na przykład można nie tylko szybko i niezawodnie minimalizować struktury molekularne, ale także przewidywać struktury stanów przejściowych i weryfikować, czy przewidywane punkty stacjonarne są w rzeczywistości minimami lub strukturą przejściową (w zależności od przypadku). Można przystąpić do obliczania ścieżki reakcji, podążając za wewnętrzną współrzędną reakcji (IRC) i określając, które reagenty i produkty są połączone daną strukturą przejściową. Gdy uzyska się pełny obraz powierzchni energii potencjalnej, można dokładnie przewidzieć energie reakcji i bariery. Można także przewidzieć szeroką gamę właściwości chemicznych.
Gaussian 16 oferuje szeroką gamę metod modelowania związków i procesów chemicznych, w tym:
Za pomocą funkcji wizualizacji GaussView można zbadać szeroki zakres wyników Gaussian:
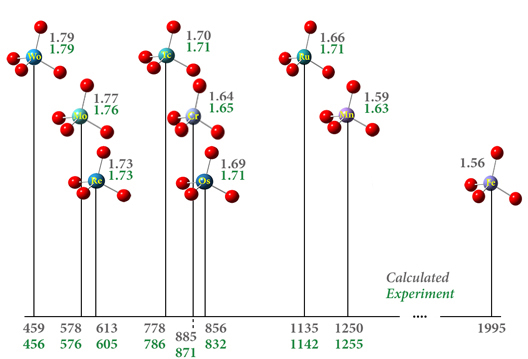
Liczba ta przedstawia przewidywane przesunięcie chemiczne w odniesieniu do pary wodnej (ppm) i długości wiązania tlen-podstawnik (Å) dla szeregu kompleksów metali przejściowych (wartości w kolorze szarym). Związki te modeluje się przy użyciu funkcjonału APFD z zestawem zasad def2-TZVPP i ECP na atomach metalu. Wyniki są w doskonałej zgodności z eksperymentem (wartości na zielono: [Kaupp95, Kaupp95a]).
Aplikacja zapewnia szeroki pakiet najbardziej zaawansowanych dostępnych możliwości modelowania. Można go używać do badania rzeczywistych problemów chemicznych w całej ich złożoności, nawet na skromnym sprzęcie komputerowym.
Co wyróżnia Gaussian 16 spośród innych programów?
- Gaussian 16 tworzy dokładne, niezawodne i kompletne modele bez konieczności chodzenia na skróty.
- Szeroka gama metod sprawia, że Gaussian 16 ma zastosowanie w szerokim zakresie warunków chemicznych, rozmiarów problemów i związków.
- Gaussian 16 zapewnia najnowocześniejszą wydajność w środowiskach jednoprocesorowych, wieloprocesorowych i wielordzeniowych, klastrach/sieciach i środowiskach obliczeniowych GPU.
- Konfigurowanie obliczeń jest proste i jednoznaczne, a nawet złożone techniki są w pełni zautomatyzowane. Elastyczne i łatwe w użyciu opcje zapewniają w razie potrzeby pełną kontrolę nad szczegółami obliczeń.
- Wyniki obliczeń prezentowane są w naturalnej i intuicyjnej formie graficznej za pomocą GaussView 6.
Podstawowe możliwości
Wychodząc od podstawowych praw mechaniki kwantowej, Gaussian 16 przewiduje energie, struktury molekularne, częstotliwości wibracyjne i właściwości molekularne związków oraz reakcje w różnorodnych środowiskach chemicznych. Modele Gaussian 16 można zastosować zarówno do stabilnych gatunków, jak i związków, które są trudne lub niemożliwe do zaobserwowania eksperymentalnie, czy to ze względu na ich naturę (np. Toksyczność, palność, radioaktywność), czy też ich wrodzoną ulotność (np. krótkotrwałe półprodukty i struktury przejściowe ).Dzięki Gaussian 16 można dokładnie zbadać problemy chemiczne. Na przykład można nie tylko szybko i niezawodnie minimalizować struktury molekularne, ale także przewidywać struktury stanów przejściowych i weryfikować, czy przewidywane punkty stacjonarne są w rzeczywistości minimami lub strukturą przejściową (w zależności od przypadku). Można przystąpić do obliczania ścieżki reakcji, podążając za wewnętrzną współrzędną reakcji (IRC) i określając, które reagenty i produkty są połączone daną strukturą przejściową. Gdy uzyska się pełny obraz powierzchni energii potencjalnej, można dokładnie przewidzieć energie reakcji i bariery. Można także przewidzieć szeroką gamę właściwości chemicznych.
Gaussian 16 oferuje szeroką gamę metod modelowania związków i procesów chemicznych, w tym:
- mechanikę molekularną EGF: Amber, UFF, Dreiding
- metody półempiryczneEGF†: m.in. AM1, PM6, PM7, DFTB
- Hartree-FockEGF
- metody funkcjonału gęstości (DFT) EGF, z obsługą wielu opublikowanych funkcjonałów; dostępne są dalekosiężne i empiryczne korekty dyspersji, jeśli zostały zdefiniowane
- kompletne samospójne pole przestrzeni aktywnej (CASSCF)EGF, w tym obsługa RAS i optymalizacje przecięć stożkowych
- teoria zaburzeń Møllera-Plesseta: MP2EGF, MP3EG, MP4(SDQ)EG, MP4(SDTQ)E, MP5E
- klaster sprzężony: CCDEG, CCSDEG, CCSD(T)E
- Brueckner gra podwójnie: BDEG, BD(T)E
- zewnętrzna funkcja wartościowości Greena (OVGF): potencjały jonizacyjne i powinowactwa elektronowe
- modele energetyczne o wysokiej dokładności: serie G1-G4, CBS i W1, wszystkie z wariantami
- metody stanu wzbudzonego: TD-DFTEGF, EOM-CCSDEG i SAC-CIEG
- energie; gradienty analityczne; częstotliwości analityczne; ponownie zaimplementowano częstotliwości analityczne.
Za pomocą funkcji wizualizacji GaussView można zbadać szeroki zakres wyników Gaussian:
- adnotacje cząsteczek i/lub zabarwienie specyficzne dla właściwości: np. ładunki atomowe, porządki wiązań, przesunięcia chemiczne NMR
- wykresy, w tym widma NMR, wibracyjne i wibrotyczne
- powierzchnie lub kontury: np. orbitale molekularne, gęstość elektronów, gęstość spinu. Właściwości takie jak potencjał elektrostatyczny można zwizualizować jako kolorową powierzchnię gęstości.
- animacje: np. tryby normalne, ścieżki IRC, optymalizacje geometrii
Właściwości molekularne w Gaussian 16
- sprzęgło antyferromagnetyczne
- ładunki atomowe
- ΔG solwatacji
- moment dipolowy
- powinowactwa elektronowe
- gęstość elektronów
- elektroniczny dichroizm kołowy (ECD)
- potencjał elektrostatyczny
- ładunki elektrostatyczne powstałe na bazie potencjału
- kształt elektronicznego pasma przejściowego
- energie o wysokiej dokładności
- nadsubtelne stałe sprzężenia (anizotropowe)
- nadsubtelne tensory widm (w tym tensory g)
- potencjały jonizacji
- widma IR i Ramana
- przedrezonansowe widma Ramana
- rezonansowe widma Ramana
- rrbitale molekularne
- momenty wielobiegunowe
- ekranowanie NMR i przesunięcia chemicznego
- stałe sprzężenia spin-spin NMR
- skręcalność optyczna (ORD)
- polaryzacyjność/hyperpolaryzacja
- aktywność optyczna Ramana (ROA)
- analiza termochemiczna
- widma UV/widzialne
- sprzęgło wibracyjno-obrotowe
- wibracyjny dichroizm kołowy (VCD)
- widma wibracyjne (absorpcyjne i emisyjne).
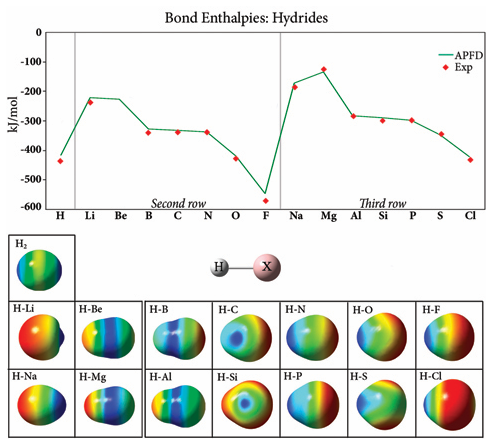
Entalpie wiązań i potencjały elektrostatyczne w wodorkach
Ten wykres przedstawia siłę wiązania w związkach wodorkowych drugiego i trzeciego rzędu (eksperyment: [CRC00]), która generalnie wzrasta w całym układzie okresowym, przy czym najsilniejsze wiązanie występuje w elemencie tuż przed gazem szlachetnym. Działka ma podobny ogólny kształt dla obu rzędów, ale wartości dla trzeciego rzędu są wyższe, ze względu na dodatkowe osłonięcie od jądra przez wypełnioną drugą powłokę. Obrazy pokazują potencjał elektrostatyczny każdego związku odwzorowany na powierzchni izodensyjności. Powierzchnia H2 ilustruje kowalencyjny charakter tego wiązania; wiązania w innych związkach wodorkowych są jonowe. Ujemny potencjał elektrostatyczny (czerwony) jest zlokalizowany na atomie wodoru na początku każdego rzędu i przesuwa się do podstawnika wraz ze wzrostem liczby atomowej w rzędzie. Zatem siła wiązań wodorkowych rośnie w okresie (wiersz) i maleje wraz ze spadkiem grupy (kolumna) ze względu na zmiany elektroujemności.
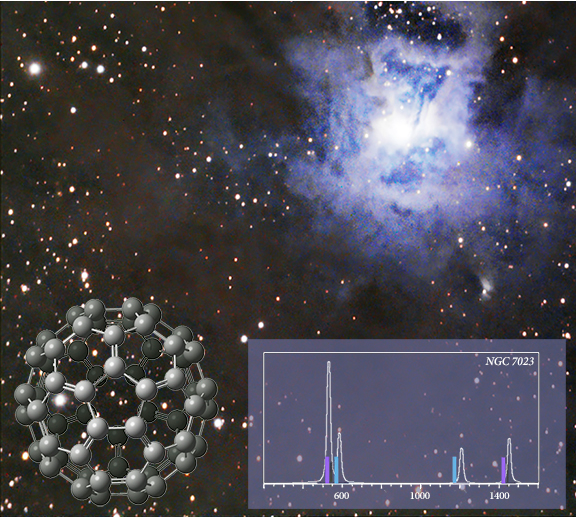
C60 w przestrzeni międzygwiezdnej
C60 wykryto w obserwacjach w podczerwieni mgławicy Irys (NGC 7023) w 2004 roku [Werner04, Sellgren10]. Wstawiony wykres pokazuje lokalizacje pików zidentyfikowane na podstawie danych (pełne słupki) nałożone na widmo przewidywane przez chemię modelu APFD/6-311+G(2d,p). Najsilniejsze piki (fioletowe) różnią się od laboratoryjnego widma IR o 0,03-0,06 µm.
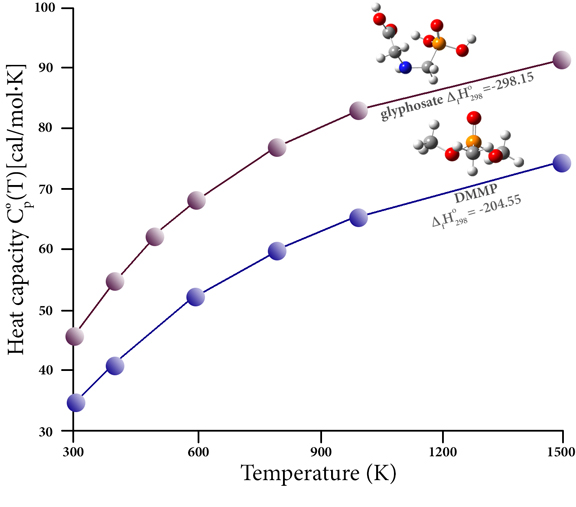
Termochemia pestycydów fosforoorganicznych
Związki fosforoorganiczne są powszechnie stosowane jako pestycydy (między innymi). Związki te niekorzystnie wpływają na zdrowie człowieka, zarówno ze względu na ich wrodzoną toksyczność, jak i na działanie szkodliwych produktów powstających podczas spalania (np. w wyniku spalania wcześniej poddanego obróbce materiału roślinnego). Rozkład tych związków jest trudny do zbadania eksperymentalnego; dane termochemiczne na ich temat są skąpe. Jednakże bardzo dokładne prognozy termochemiczne mogą wypełnić tę lukę i umożliwić badanie stabilności termicznej odpowiednich związków i produktów spalania. Na przykład ten wykres przedstawia pojemność cieplną w funkcji temperatury dla dwóch takich związków: glifosatu będącego pestycydem i łagodniejszego związku zmniejszającego palność, metylofosfonianu dimetylu (DMMP). Podaje także ciepło ich powstawania (kcal/mol), jak przewidywano na podstawie obliczeń CBS-QB3 Khalfy i współpracowników [Khalfa15]. W ich artykule przedstawiono wyniki obliczeń dla dużej liczby trójwartościowych i pięciowartościowych związków fosforu, co umożliwia zaproponowanie 83 oryginalnych grup do zastosowania w półempirycznej metodzie wkładu grupowego Bensona, a tym samym ocenę właściwości termochemicznych niektórych powszechnych związków fosforu. pestycydy, herbicydy i związki pokrewne.Przeglądaj egzotyczne części układu okresowego
Gaussian zapewnia funkcje modelowania ciężkich pierwiastków, dla których istotne są efekty relatywistyczne. Zestawy podstawowe grup Stuttgart-Drezno i Ahlrichs + ECP są wbudowane, a ich traktowanie określonych elementów można połączyć z innymi standardowymi zestawami bazowymi za pomocą bardzo prostych specyfikacji wejściowych.Przesunięcia chemiczne 17O w kompleksach metali przejściowych
Liczba ta przedstawia przewidywane przesunięcie chemiczne w odniesieniu do pary wodnej (ppm) i długości wiązania tlen-podstawnik (Å) dla szeregu kompleksów metali przejściowych (wartości w kolorze szarym). Związki te modeluje się przy użyciu funkcjonału APFD z zestawem zasad def2-TZVPP i ECP na atomach metalu. Wyniki są w doskonałej zgodności z eksperymentem (wartości na zielono: [Kaupp95, Kaupp95a]).
Nowości w pakiecie
Nowości funkcjonalne w pakietu Gaussian 16
Wymagania aplikacji
Wymagania systemowe programu Gaussian